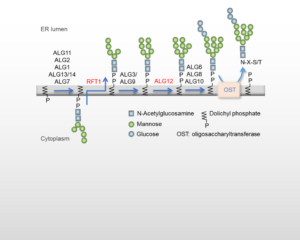
Les CDG de type I (CDG-I) sont un groupe d’environ 30 maladies métaboliques héréditaires rares affectant le processus de N-glycosylation des glycoprotéines dans le réticulum endoplasmique (Fig 1). Les CDG-I présentent un tableau clinique multisystémique complexe et souvent sévère. L’hôpital Bichat est un centre de référence pour le diagnostic du CDG-I en France. Le signe distinctif biochimique des CDG-I est la présence de glycoprotéines sériques hypoglycosylées (dont la transferrine) et, de nos jours, les origines moléculaires de la maladie sont généralement identifiées par séquençage de l’exome entier.
Un patient présente une transferrine hypoglycosylée et est porteur d’un variant homozygote de RFT1. Le radiomarquage métabolique des fibroblastes d’un patient, suivi de la caractérisation des oligosaccharides liés au dolichol au cours du processus de N-glycosylation, n’a pas révélé l’accumulation de structures Man5GlcNAc2-PP-dolichol tronqué attendue d’un RFT1-CDG mais une accumulation de Man7GlcNAc2-PP-dolichol, caractéristique d’un ALG12-CDG . La réévaluation des données NGS a révélé un variant homozygote qui modifie le deuxième nucléotide du premier intron du gène ALG12. Une insertion de 4 bases entre l’exon 1 et l’exon 2 a été trouvée dans l’ADNc, suggérant un déplacement de l’épissage de l’ARNm dans cet intron vers un nouveau site donneur GU putatif. Il s’agit de la première description d’une mutation intronique associée à une altération de l’épissage de l’ARNm ALG12 et à un très faible taux d’ARNm ALG12.