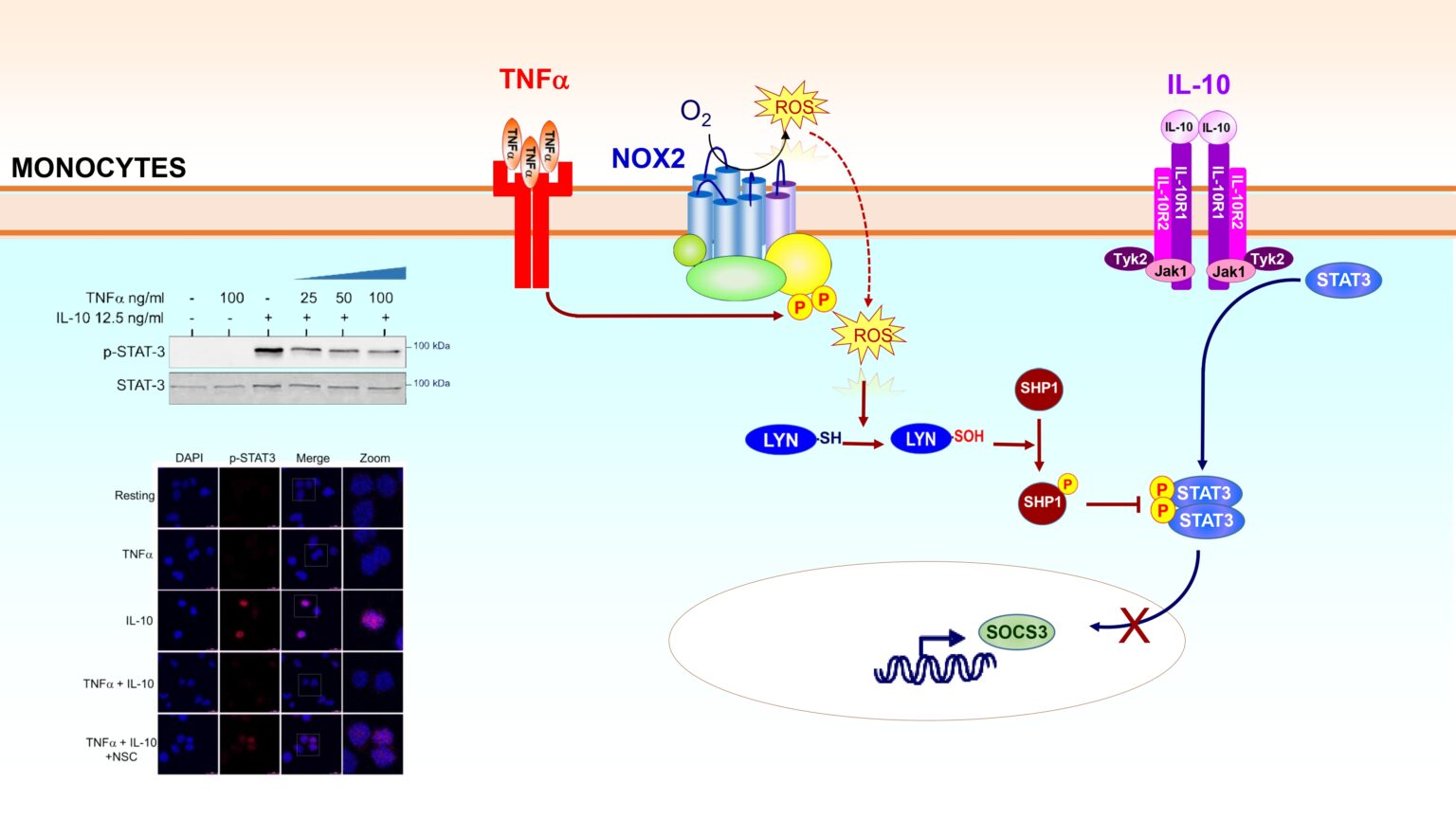


TNFa-controlled intracellular signaling pathways play a central role in the pathogenesis of inflammatory diseases such as rheumatoid arthritis (RA) and chronic inflammatory bowel disease (IBD) by promoting phagocyte inflammatory functions such as cytokine release and production of reactive oxygen species (ROS) by the NADPH oxidase NOX2.
In contrast, interleukin-10 (IL-10), a potent anti-inflammatory cytokine, inhibits phagocyte activation, making IL-10 an attractive therapeutic candidate. However, for reasons that remain unknown, IL-10-based therapies have shown little efficacy in patients with inflammatory diseases.
Our study reveals that TNFα impairs IL-10 signaling in human monocytes thereby prolonging inflammation. Mechanistically, we demonstrate that TNFα inhibits IL-10-induced STAT3 activation by activating SHP1 phosphatase via the NOX2-ROS-Lyn axis. Inhibition of STAT3 by TNFa results in decreased expression of SOCS3, a major anti-inflammatory factor induced by IL-10.
In addition, DPI, a NOX2 inhibitor, reverses the inhibitory effect of TNFa on IL-10 signaling, and H2O2, a molecule generated by NOX2, reproduces this inhibitory effect of TNFa. Finally, in vivo, pharmacological inhibition of SHP-1 protects mice against arthritis induced by anti-collagen antibodies.
These findings may explain the low efficacy of IL-10-based therapies in patients with chronic inflammatory diseases such as RA and IBD, and suggest that anti-TNFa and SHP1/2 inhibitors could improve the therapeutic use of IL-10.