Authors
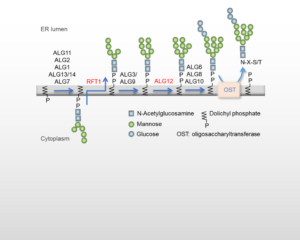
CDG type I (CDG-I) is a group of around 30 rare inherited metabolic disorders affecting the N-glycosylation of glycoproteins in the endoplasmic reticulum (Fig 1). CDG-I presents a complex and often severe multisystem clinical picture. Hôpital Bichat is a reference center for the diagnosis of CDG-I in France. The biochemical hallmark of CDG-I is the presence of hypoglycosylated serum glycoproteins (including transferrin), and nowadays the molecular origins of the disease are generally identified by whole exome sequencing.
One patient has hypoglycosylated transferrin and carries a homozygous RFT1 variant. Metabolic radiolabeling of patient fibroblasts, followed by characterization of dolichol-bound oligosaccharides during the N-glycosylation process, did not reveal the accumulation of truncated Man5GlcNAc2-PP-dolichol structures expected of an RFT1-CDG but an accumulation of Man7GlcNAc2-PP-dolichol, characteristic of an ALG12-CDG . Reassessment of the NGS data revealed a homozygous variant that modifies the second nucleotide of the first intron of the ALG12 gene. A 4-base insertion between exon 1 and exon 2 was found in the cDNA, suggesting a shift of mRNA splicing in this intron to a putative new GU donor site. This is the first description of an intronic mutation associated with altered ALG12 mRNA splicing and very low ALG12 mRNA levels.